

By: Diego A. Rodríguez Soacha
BPharm, MSc, Dr. rer. Nat. Senior Medicinal Chemist
The journey of drugs within the body underscores pharmacokinetics as a cornerstone of effective drug development. By studying the ADME (Absorption, Distribution, Metabolism, and Excretion) phases, it highlights how each step influences the safety and efficacy of a drug. Understanding the dynamic interactions of drugs with the human body allows us to push the limits of medical science, ensuring that new therapies are not only potent, but also safe and fitted to meet the diverse needs of patients.
Medications have become a commonplace tool in our homes, routinely used to prevent, manage symptoms, or cure diseases. We know the basic rules: store them in dry places, away from light, and always check their expiration dates. Yet, behind these everyday essentials lie numerous myths and misunderstandings. A common question that intrigues many is about their functionality: how do medications “know” where to act? For instance, if you have a knee pain, how does an analgesic “understand” precisely where to target its effects? This article delves into the world of pharmacokinetics to uncover how drugs navigate through the body, demystifying the journey from the medicine cabinet to the cellular level.
To understand how medications work, we need to understand two key terms: pharmacokinetics (PK) and pharmacodynamics (PD). Pharmacodynamics explains “what the drug does to our body”. For example, it describes how certain analgesics block pain signals from nerves to the brain, or how antipyretics act on the brain’s temperature-regulating center to reduce body temperature and control fever. On the other hand, pharmacokinetics deals with “the processes that our body subjects the drug to”. These successive processes, known under the acronym “ADME” (Absorption, Distribution, Metabolism, and Excretion), are dependent on the drug’s route of administration.
Before a drug can exert its effects, it must separate from its formulation components and enter the bloodstream through absorption. The process of absorption is influenced by several factors, with the route of administration being one of the most important.1 Except for intravenous (IV) administration, where the drug is directly delivered into the bloodstream thereby bypassing the absorption step, all other routes require the drug to be absorbed from the site of administration.1 Certain routes can lead to a quicker onset of action, primarily due to faster absorption rates.2 Similarly, each route requires that the drug formulation includes different components, known as excipients, to ensure that the drug is compatible with its biological environment.
To understand why certain drug formulations are specific to the intramuscular (IM) route and not suitable for intravenous (IV) administration, we must consider the unique characteristics of each pathway. Intramuscular injections involve depositing the drug into the muscle tissue, where it is slowly absorbed into the bloodstream.2,3 This process is influenced by the formulation’s interaction with the muscle’s extracellular matrix (ECM), a network that supports the gradual release and absorption of the drug.3
However, IV administration bypasses this absorption phase by directly entering the bloodstream, delivering immediate systemic effects. Certain IM-specific formulations contain excipients that optimize the drug’s release from the muscle tissue but can cause complications if introduced directly into the bloodstream.4 For instance, these excipients may induce hemolysis (destruction of red blood cells) if administered intravenously. This distinction highlights why a formulation designed for IM use, which might include agents to control the drug’s release speed or reduce local irritation, could lead to severe adverse reactions if misused in an IV context.4
On the other hand, oral administration is the preferred route for many medications due to its non-invasive nature, making it particularly user-friendly for most patients. The gastrointestinal (GI) tract, particularly the stomach and small intestine, plays a vital role in drug absorption. The stomach, with its relatively small absorptive area of about 1 m², initiates digestion, but it is the small intestine that is crucial for drug absorption. This part of the GI tract boasts a vast absorptive surface of approximately 200 m² due to the presence of microvilli, enhancing its ability to efficiently transport nutrients and drugs into the bloodstream.2 The lining of the intestine includes a variety of cells such as goblet cells, which produce mucus, and absorptive cells, essential for transferring substances from the GI lumen to the blood.5
However, the effectiveness of oral administration can be influenced by the varying pH levels within the GI tract. While these pH variations can aid in the dissolution and absorption of certain drugs, they can also pose risks by potentially degrading sensitive compounds.6 Therefore, proper formulation of oral medications must account for these environmental conditions to prevent molecule degradation and ensure therapeutic efficacy.
Now that the drug has been absorbed and entered the bloodstream, the next phase of its journey is distribution, which ensures that the drug reaches its site of action and can exert its biological effect. Distribution is a key pharmacokinetic process where the drug disperses throughout the body, to and from the blood and various tissues or organs.2 This phase is critical in determining how effectively a drug can achieve and maintain the concentrations necessary for therapeutic action at the target sites.
Measuring key pharmacokinetic parameters, such as “Volume of Distribution”, which provides information of how extensively a drug is distributed outside the vascular system,7 helps understand the nature of each molecule within the organism. Several factors affect drug distribution2,7 including:
- Blood flow to tissues: Organs with higher blood perfusion rates, such as the liver and kidneys, generally receive more of the drug quickly.
- Tissue binding: Drugs may bind to tissue components, which can sequester the drug away from the bloodstream.
- Membrane permeability: The drug’s ability to cross cell membranes affects its distribution, influenced by the drug’s lipophilicity and molecular size.
- Physiological barriers: Structures like the blood-brain barrier (BBB) can restrict drug access to specific tissues, impacting the overall distribution and effectiveness.
Focusing on drugs aimed at treating neurodegenerative disorders like Alzheimer’s disease (AD), where the involved receptors are mainly expressed in the brain, crossing biological barriers, particularly the blood-brain barrier (BBB), presents a significant challenge. The BBB is a highly selective barrier, composed of endothelial cells that line the cerebral blood vessels, tightly joined together by complex tight junctions.8 This barrier regulates the entry of substances into the brain, ensuring that the central nervous system’s environment remains stable and protected from potentially harmful substances circulating in the blood.8,9
Drugs cross the BBB through mechanisms such as passive diffusion, active transport, and receptor-mediated transcytosis 10
- Passive diffusion: Small, lipophilic (fat-soluble) molecules can often pass through the BBB via passive diffusion. This method depends on the drug’s ability to dissolve in the cell membrane, which is largely composed of lipids.
- Active transport: Specific transport proteins can carry certain molecules across the BBB, including glucose and amino acids. Some drugs are designed to mimic these molecules to utilize these transport systems.
- Receptor-mediated transcytosis: Some larger molecules, including certain therapeutic proteins, can be transported across the BBB by binding to specific receptors on the surface of endothelial cells, which then transport the drug across the cell and release it on the other side.
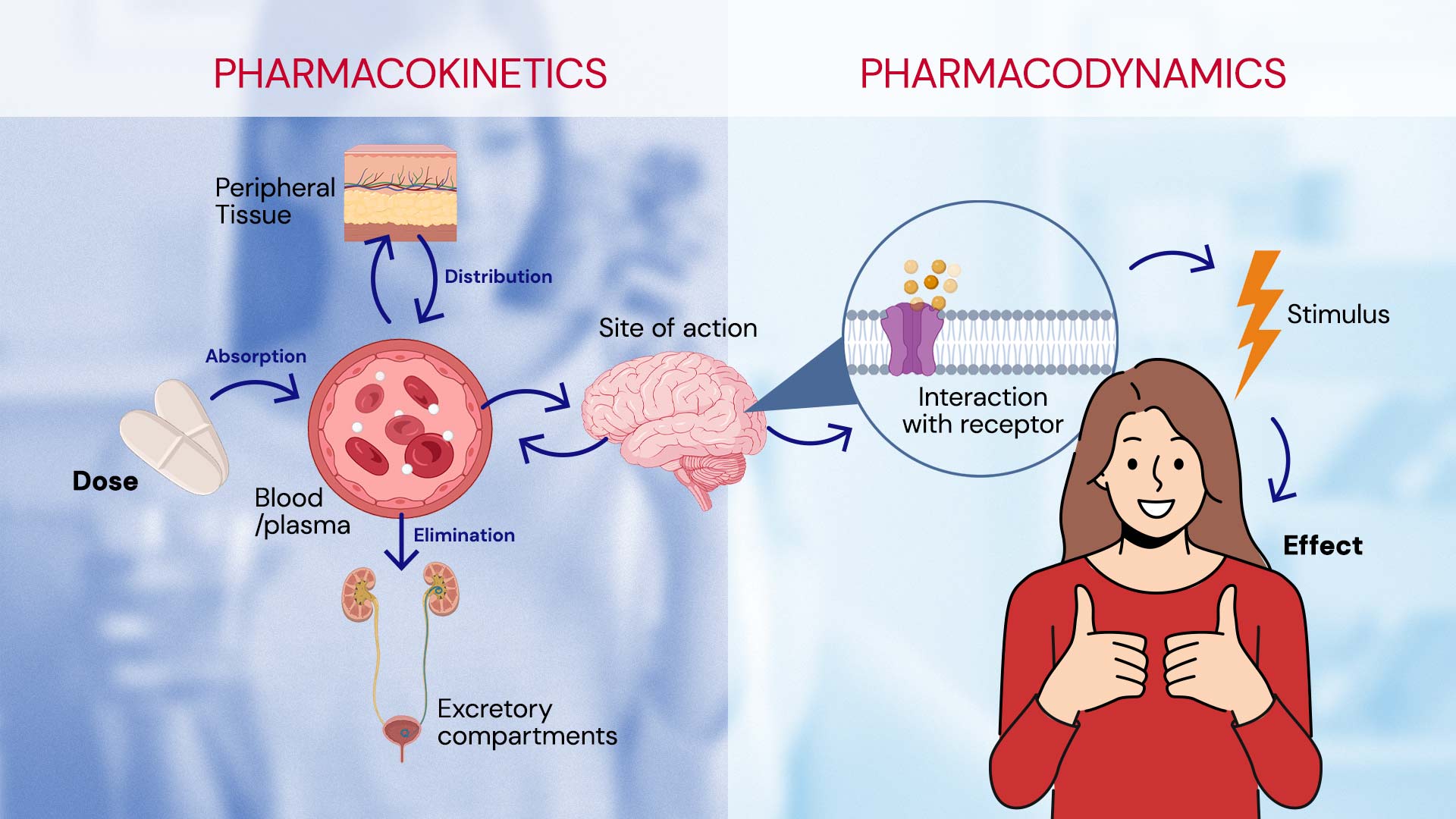
Once the drug reaches its target and exerts its biological effect, it is time for it to leave the body. While the process of metabolism can occur at any point, even before absorption, ideally, the drug should be metabolized and eliminated once its therapeutic effect is no longer needed.2 Metabolism involves the body’s processes that modify the drug. This often includes making the drug more polar through chemical changes, enabling it to be excreted.11 This process can occur in various organs, primarily in the liver, which plays a crucial role in drug metabolism.11,12 Drugs and other xenobiotics undergo phase I and phase II metabolism: 2,13
- Phase I metabolism involves modifications such as oxidation, reduction, or hydrolysis. These reactions, often mediated by enzymes like the cytochrome P450 family, can result in a metabolite that is either active, inactive, or sometimes more active than the original drug.
- Phase II metabolism involves conjugation where the drug or its phase I metabolites are coupled with another substance like glucuronic acid, sulfate, or glycine, making them more water-soluble and easier to excrete.
Metabolites vary in their activity; some are inactive, while others can retain the same activity level or even be more active than the parent drug.11-13, In some cases, prodrugs are used, which are inactive in their administered form and require metabolic activation to become effective.11
The final phase of a drug’s long journey through the body is excretion. Excretion is the process by which drugs and their metabolites are physically removed from the body, primarily through the kidneys via urine, but also through bile, feces, sweat, and exhaled air. If drugs or their metabolites are not removed from the body, they can accumulate in various tissues, causing toxicity and adverse effects. 1 The main route of excretion often depends on the physical and chemical properties of the substance:14
- Renal excretion: This is the most common pathway for the elimination of drugs and involves filtration, secretion, and reabsorption processes within the kidneys. Drugs that are small, water-soluble, and non-bound to plasma proteins are preferentially excreted by the kidneys.
- Biliary and fecal excretion: Drugs that are large, have high molecular weight, or are conjugated to large molecules in the liver are often excreted into the bile and then eliminated in the feces. Some drugs undergo enterohepatic recirculation, where they are reabsorbed and then secreted multiple times, prolonging their presence in the body.14,15
By measuring the variables that affect drug excretion, we can define the “clearance” parameter, which describes the volume of plasma from which a drug is completely cleared per unit of time.16 It is a critical measure of the efficiency with which a drug is cleared from the body and depends on both the drug’s ability to reach the organs of elimination (like the kidneys or liver) and the organ’s ability to process and eliminate the drug.15,16 This parameter directly affects how quickly a drug is removed from the body, influencing both the drug’s efficacy and safety.16
The drug’s journey from absorption to excretion reflects the intricate biological processes shaping its efficacy and safety. For us at IGC Pharma, a detailed understanding and evaluation of pharmacokinetic parameters are not mere academic exercises; they are essential practices that guide the development of our new molecules. By carefully evaluating how drugs are absorbed, distributed, metabolized, and excreted, we can design medicines that are not only effective, but also safer for the patient. This meticulous approach ensures that we continue to advance our ability to offer more targeted, efficient, and tolerable treatments. As we move forward, the insights gained from pharmacokinetics will continue to be contributory in our quest to innovate and enhance patient care, making every effort to turn potential risks into predictable outcomes.
- Robertson, D. (2017). Introduction to pharmacokinetics. Nurse Prescribing, 15(3), 146-148.
- Benedetti, M. S., Whomsley, R., Poggesi, I., Cawello, W., Mathy, F. X., Delporte, M. L., … & Watelet, J. B. (2009). Drug metabolism and pharmacokinetics. Drug metabolism reviews, 41(3), 344-390
- McCartan, A. J., Curran, D. W., & Mrsny, R. J. (2021). Evaluating parameters affecting drug fate at the intramuscular injection site. Journal of Controlled Release, 336, 322-335.
- Amin, K., & Dannenfelser, R. M. (2006). In vitro hemolysis: guidance for the pharmaceutical scientist. Journal of pharmaceutical sciences, 95(6), 1173-1176.
- Murakami, T. (2017). Absorption sites of orally administered drugs in the small intestine. Expert opinion on drug discovery, 12(12), 1219-1232.
- Kosugi, Y., Yamamoto, S., Sano, N., Furuta, A., Igari, T., Fujioka, Y., & Amano, N. (2015). Evaluation of acid tolerance of drugs using rats and dogs controlled for gastric acid secretion. Journal of Pharmaceutical Sciences, 104(9), 2887-2893.
- Gopinath, A., & Wilson, M. (2020). Factors affecting drug absorption and distribution. Anaesthesia & Intensive Care Medicine, 21(5), 223-228.
- Pandit, R., Chen, L., & Götz, J. (2020). The blood-brain barrier: Physiology and strategies for drug delivery. Advanced drug delivery reviews, 165, 1-14.
- Loryan, I., Hammarlund-Udenaes, M., & Syvänen, S. (2020). Brain distribution of drugs: Pharmacokinetic considerations. In Physiology, Pharmacology and Pathology of the Blood-Brain Barrier (pp. 121-150). Cham: Springer International Publishing.
- Neumaier, F., Zlatopolskiy, B. D., & Neumaier, B. (2021). Drug penetration into the central nervous system: pharmacokinetic concepts and in vitro model systems. Pharmaceutics, 13(10), 1542.
- Zhang, Z., & Tang, W. (2018). Drug metabolism in drug discovery and development. Acta Pharmaceutica Sinica B, 8(5), 721-732.
- Almazroo, O. A., Miah, M. K., & Venkataramanan, R. (2017). Drug metabolism in the liver. Clinics in liver disease, 21(1), 1-20.
- Iyanagi, T. (2007). Molecular mechanism of phase I and phase II drug‐metabolizing enzymes: implications for detoxification. International review of cytology, 260, 35-112.
- Boussery, K., Belpaire, F. M., & Van de Voorde, J. (2008). Physiological aspects determining the pharmacokinetic properties of drugs. In The practice of medicinal chemistry (pp. 635-654). Academic Press.
- Malik, M. Y., Jaiswal, S., Sharma, A., Shukla, M., & Lal, J. (2016). Role of enterohepatic recirculation in drug disposition: cooperation and complications. Drug metabolism reviews, 48(2), 281-327.
- Toutain, P. L., & Bousquet‐mélou, A. (2004). Plasma clearance. Journal of veterinary pharmacology and therapeutics, 27(6), 415-425.




